


Repurposing FDA‑Approved Drugs to Inhibit SARS‑CoV‑2 Main Protease: A Virtual Screening Analysis
Targeting the SARS‑CoV‑2 Main Protease
Developing a novel antiviral is both expensive and time‑consuming. In the midst of a global pandemic, the urgent need is to identify drugs that can halt viral replication rapidly. Drug repurposing—leveraging medicines already proven safe in humans—offers a promising shortcut. While single drugs may provide modest benefits, strategic combinations that attack multiple viral proteins, as in the case of HIV therapy, could yield substantial clinical gains. The key question: which combinations will be most effective against SARS‑CoV‑2?
Our study focuses on the protease’s active‑site architecture, comparing it to the well‑characterized SARS‑CoV protease bound to micromolar inhibitors. This structural insight guides the design of potent inhibitors for SARS‑CoV‑2.
We then performed a virtual screening of an FDA‑approved drug library to identify molecules predicted to bind the protease and to evaluate their potential in combination therapies.
SARS‑CoV‑2 Proteins
The rapid sequencing of viral genomes from patients provided the sequences of numerous potential drug targets. Many of these proteins share high similarity with SARS‑CoV counterparts, enabling initial homology models. As experimental structures (X‑ray, Cryo‑EM) become available, they refine our understanding of druggable sites.
Among coronaviruses, the main protease (Mpro, also known as 3CL Protease) is a leading drug target. Alongside papain‑like proteases, Mpro cleaves the polyprotein 1ab at 11 defined sites, a process essential for viral maturation.
Inhibiting Mpro blocks replication. Because no human protease shares its cleavage specificity, inhibitors are less likely to exhibit off‑target toxicity.
Structure‑Based Design for Repurposing: Mpro
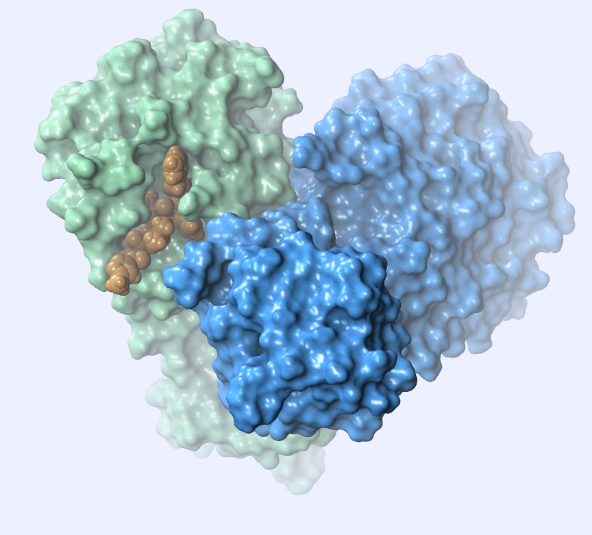
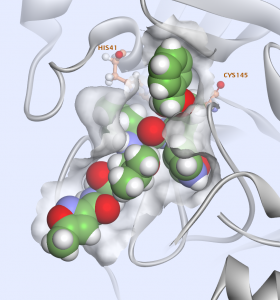
Mpro functions as a homodimer (Fig. 2), each subunit containing a catalytic His41/Cys145 dyad.
The SARS‑CoV‑2 protease shares 96.1% identity and 99% similarity with its SARS‑CoV ancestor. Only one residue differs in the active site: position 46 is Serine instead of Alanine (Fig. 3).
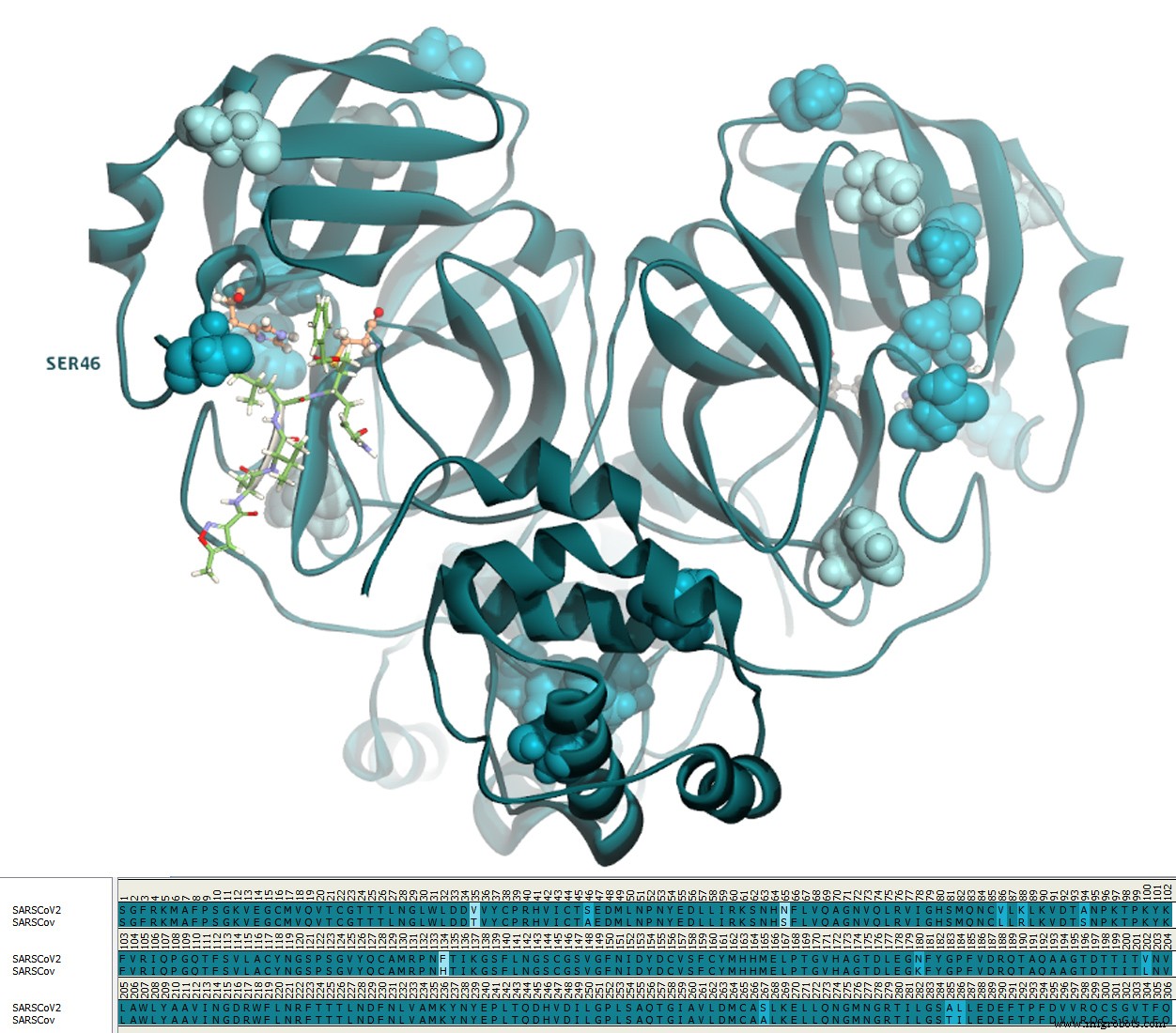
Numerous SARS‑CoV Mpro structures complexed with micromolar inhibitors are available; the most potent ligands are covalent. The active site is subdivided into four subsites (S1′, S1, S2, S4) that accommodate diverse chemotypes.
In high‑affinity complexes (PDB 2ZU4 and 2GX4; Ki = 0.038 µM and 0.053 µM), the cysteine 145 thiol forms a covalent bond with the inhibitor, underscoring the importance of covalent engagement for strong binding.
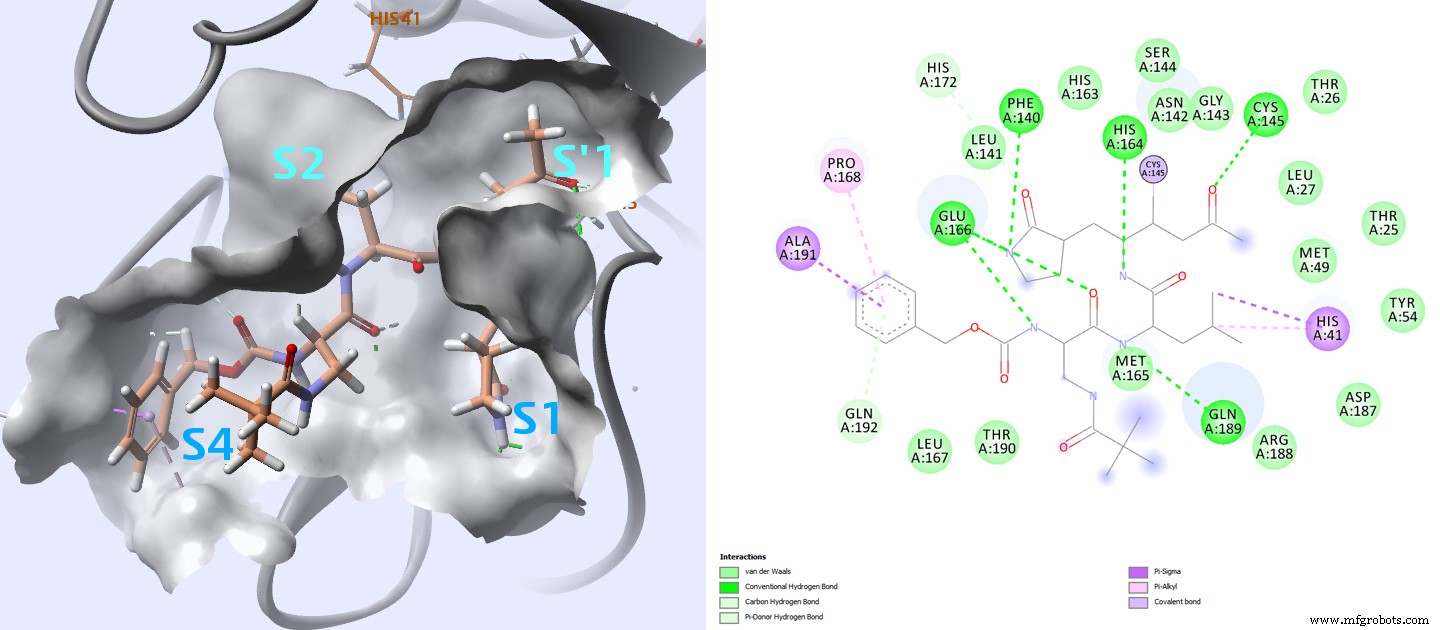
Detailed analysis of these interactions informs the key contacts to monitor in docking studies.
Virtual Screening Workflow
Using the 2.5 Å crystal structure of the SARS‑CoV‑2 protease dimer bound to the covalent inhibitor N3 (PDB 6LU7) released in February, we conducted a systematic virtual screen. N3 occupies the substrate‑binding pocket in an extended conformation (Fig. 5).
Our library comprised 2,684 FDA‑approved drugs with molecular weight < 800 Da. Docking was performed with GOLD (CCDC), followed by binding‑free‑energy calculations using CHARMM/GBMV. Each pose underwent in‑situ ligand minimization (14 Å sphere) and ligand‑entropy estimation. Many top‑scoring compounds were known HIV/HCV protease inhibitors (Fig. 6).
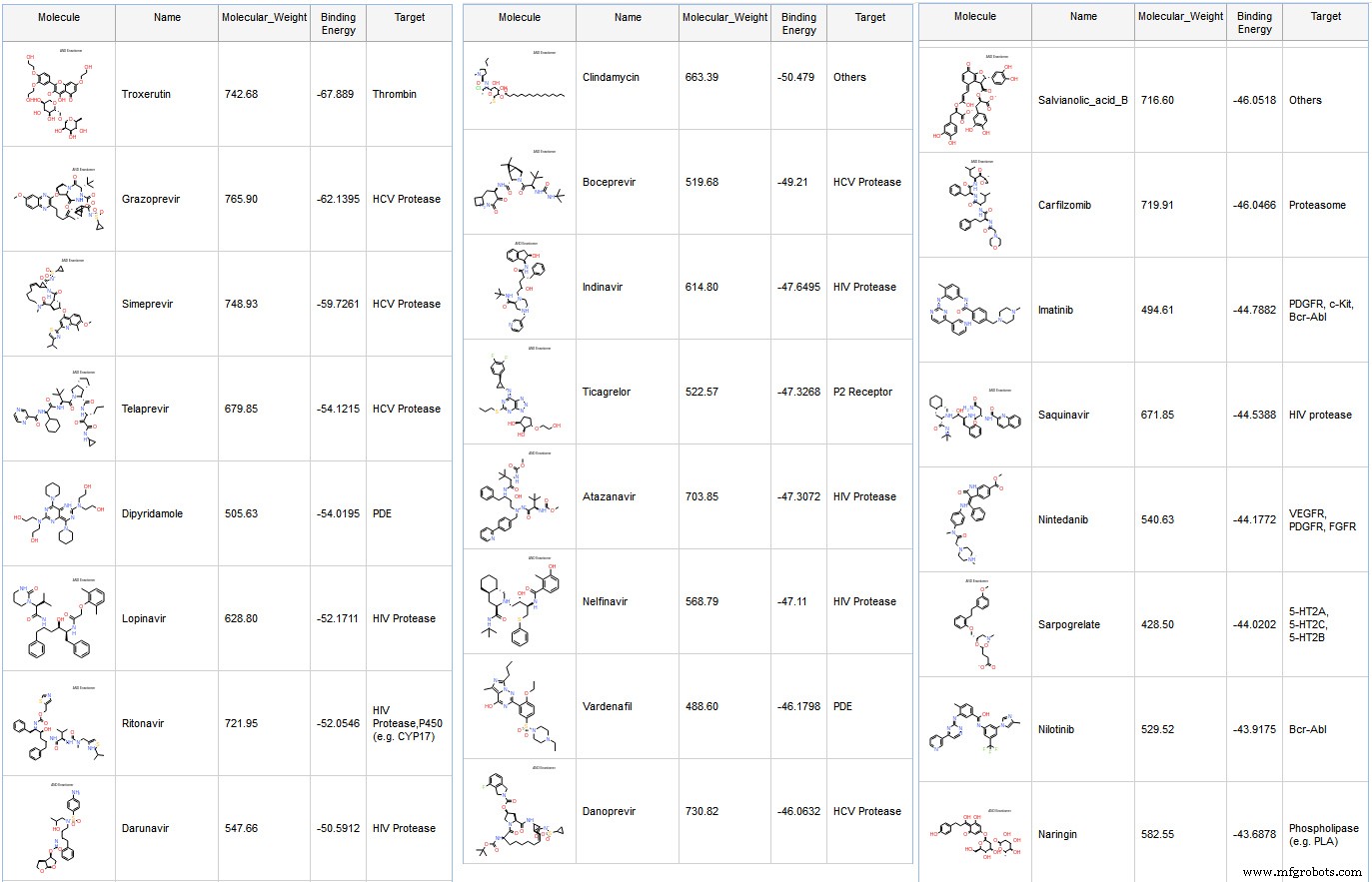
The best candidate, Troxerutin—a flavonoid—scored highly. Flavonoids have documented protease‑inhibitory activity; recent IC₅₀ values for herbacetin, rhoifolin, and pectolinarin against SARS‑CoV were 33.17, 27.45, and 37.78 µM, respectively.
Dipyridamole (Fig. 7) ranked fifth. A recent preprint reports its ability to suppress HCoV‑19 replication with an EC₅₀ of 100 nM in vitro.
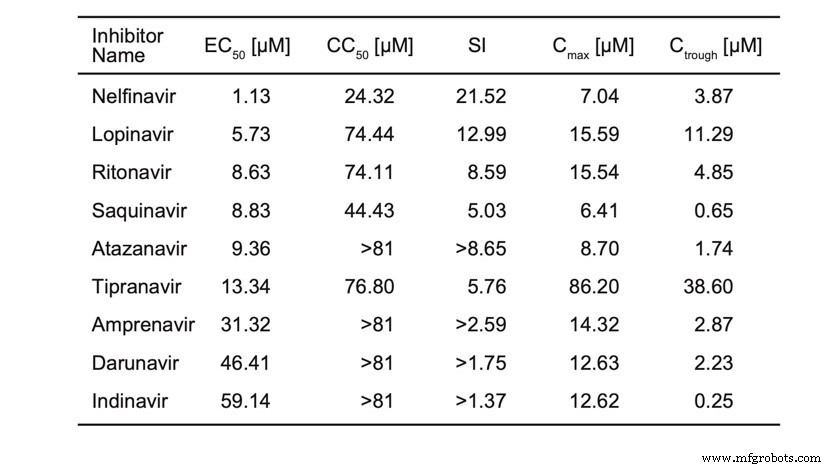
Preprint studies of HIV protease inhibitors further support activity against SARS‑CoV‑2. Nelfinavir, for example, inhibited viral replication in Vero E6 cells.
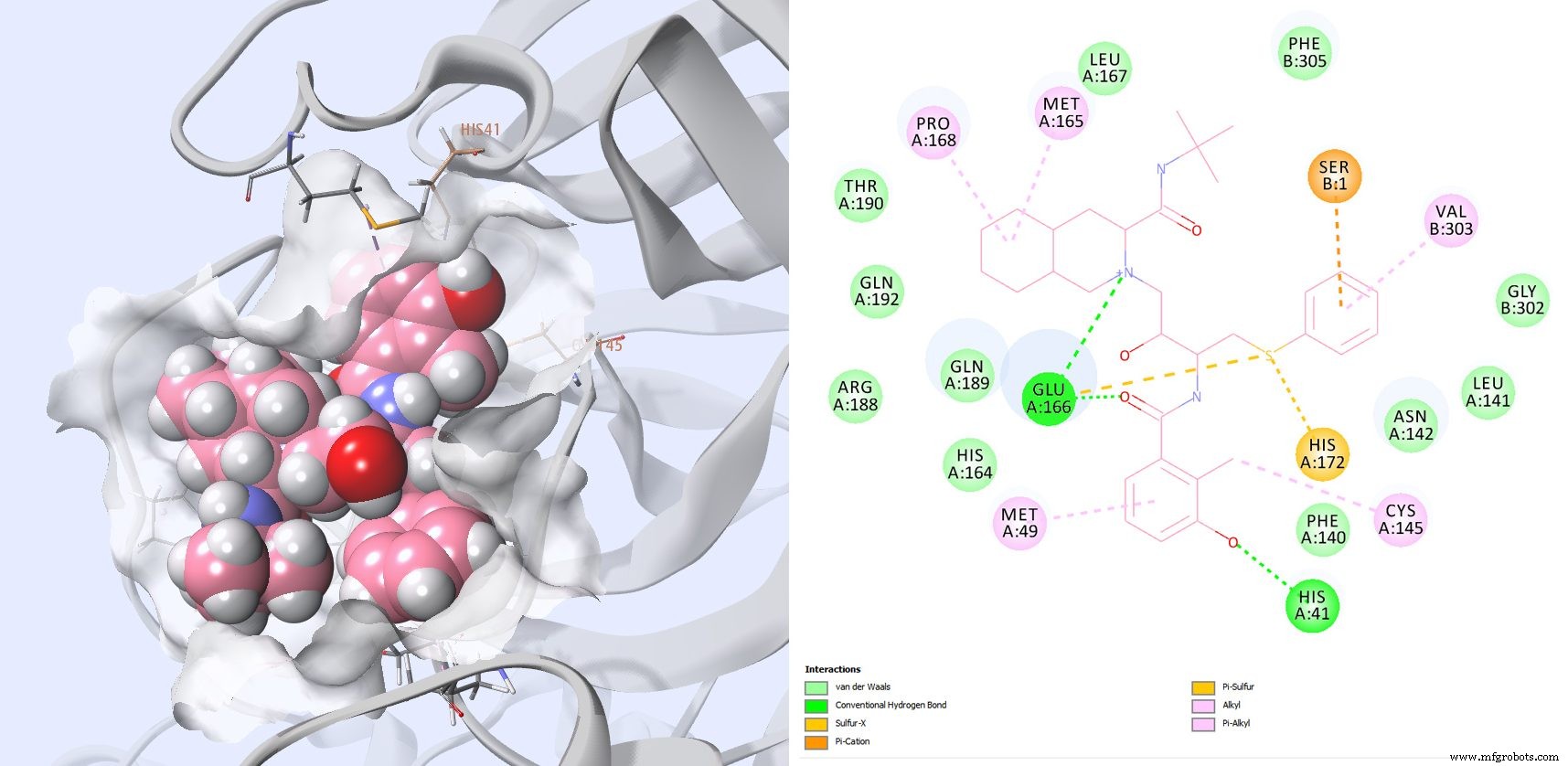
Ritonavir and Lopinavir, both interacting with catalytic residues and additional key side chains (GLU166, PRO168, GLN189), are under clinical investigation. However, IC₅₀ data for these agents against SARS‑CoV‑2 remain unavailable.
Atazanavir also shows activity in infected cells, reducing viral replication and pro‑inflammatory cytokine production. A newly released structure (PDB 6W63) featuring inhibitor X77 shares a similar binding‑site orientation to 6LU7; re‑docking confirmed comparable predictions.
Implications and Future Directions
With four subsites, the Mpro pocket can accommodate diverse ligands with moderate affinity. Virtual screening thus serves as a powerful tool to pinpoint candidate inhibitors and elucidate their binding modes.
These computational hypotheses require experimental validation—IC₅₀ measurements, covalent docking, and pharmacokinetic profiling—to develop clinically viable therapeutics. Covalent inhibitors, in particular, often exhibit superior potency and prolonged action, making them attractive leads.
Ultimately, this work provides a rapid, structure‑guided roadmap for repurposing existing drugs against COVID‑19, facilitating the urgent translation from bench to bedside.
We thank the Cambridge Crystallographic Data Centre for access to GOLD; its proven efficacy in virtual screening and lead optimization underpins this study. An interface to GOLD is available within Discovery Studio.
Biologics
- Nicotine Patch: Science, Manufacturing, and Future Innovations
- Pharmacophore‑Guided Virtual Screening Enhances Drug Repurposing for COVID‑19
- Accelerating Drug Discovery with AI‑Powered Generative Therapeutics Design
- Leveraging Molecular Modeling to Uncover Novel Therapeutic Targets for SARS‑CoV‑2
- Decoding SARS‑CoV‑2 Genomes: DNA & Antibody Tests for COVID‑19
- Unraveling the Origins of SARS‑CoV‑2: A Genomic Perspective
- China Moves Toward a U.S.-Style Patent‑Linkage System for Drugs
- Unified COVID-19 Test: Detect, Track Variants & Co-Infections in One Procedure
- Revolutionary Smartwatch Monitors Real-Time Medication Levels
- RapidPlex Sensor: 5 Essential Questions About SARS-CoV-2 At‑Home Testing