


Methane Adsorption on MoX₂ (S, Se, Te) Monolayers: Insights from DFT
Abstract
Using density‑functional theory, we examined how CH₄ interacts with monolayer MoX₂ (X = S, Se, Te) containing X‑vacancies, Mo‑vacancies, and divacancies. The study shows that defect engineering markedly improves methane adsorption. Among the defect types, the divacancy in MoTe₂ (MV_D) delivers the strongest adsorption, with the largest charge transfer and the shortest adsorption distance. These findings highlight MV_D(MoTe₂) as a promising platform for methane sensing devices.
Introduction
Methane (CH₄) is a colorless, tasteless, and non‑toxic gas, yet high concentrations can displace oxygen and pose health risks. When methane levels exceed 25–30 % of the atmosphere, symptoms such as headaches, dizziness, and rapid breathing may appear. Two‑dimensional transition‑metal dichalcogenides (TMDs) such as MoX₂ (X = S, Se, Te) have attracted attention for their intrinsic band gaps and potential in gas‑sensing applications. Defects—especially vacancies—alter the electronic properties of TMDs, influencing their interaction with adsorbates. Here we use first‑principles calculations to explore methane adsorption on pristine and defected MoX₂ monolayers, aiming to identify the most effective sensor material.
Method and Theory
A 4 × 4 supercell of MoX₂ (32 X atoms, 16 Mo atoms) was constructed and CH₄ was placed above it in Materials Studio. All calculations were performed with DMol³, employing the PBE functional and a 6 × 6 × 1 k‑point mesh. The cutoff energy was set to 340 eV, with self‑consistency convergence at 10⁻⁵ eV. Atomic positions were relaxed until forces were below 0.03 eV/Å. Adsorption energies were evaluated via
\[E_{ad}=E_{MoX₂+CH₄}-\left(E_{MoX₂}+E_{CH₄}\right)\]
and charge transfer was analyzed using Mulliken population analysis.
Results and Discussion
Geometric and Electronic Structures of MoX₂
Optimized Mo–X bond lengths are 2.426 Å (Mo–S), 2.560 Å (Mo–Se), and 2.759 Å (Mo–Te), matching experimental values (2.410 Å, 2.570 Å, 2.764 Å). Introducing vacancies shortens the neighboring Mo–X bonds, reflecting stronger local interactions.
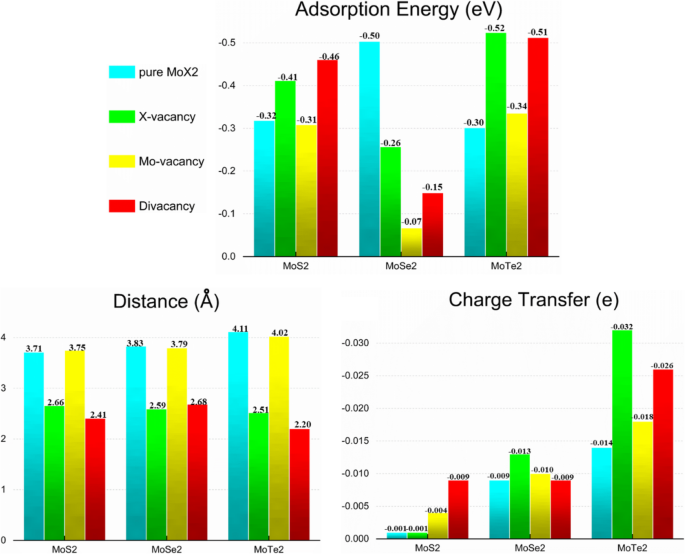
Adsorption Energies, Distances, and Charge Transfer
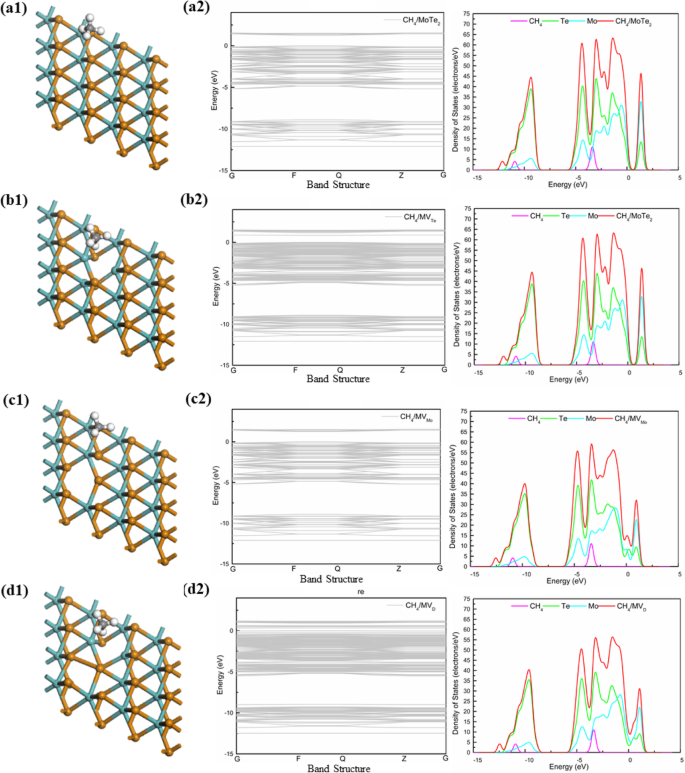
Prior to adsorption, CH₄ is 3.6 Å above the surface. Adsorption energies, corrected for van der Waals interactions, range from –0.31 eV to –0.52 eV for MoS₂, –0.07 eV to –0.50 eV for MoSe₂, and –0.30 eV to –0.52 eV for MoTe₂. The strongest binding occurs on the divacancy in MoTe₂, with a CH₄–surface distance of 2.20 Å and a charge transfer of –0.026 e. Charge transfer values indicate that CH₄ acts as an electron acceptor in all cases.
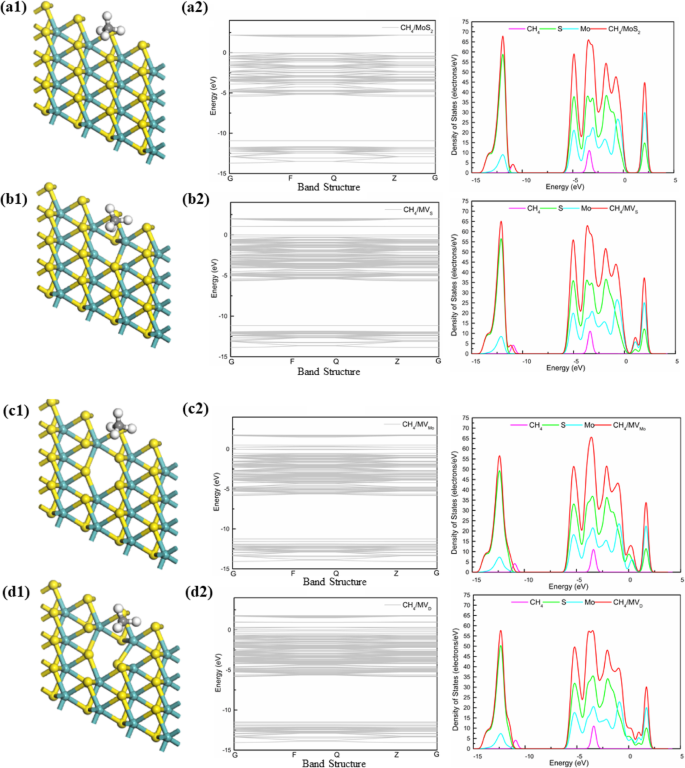
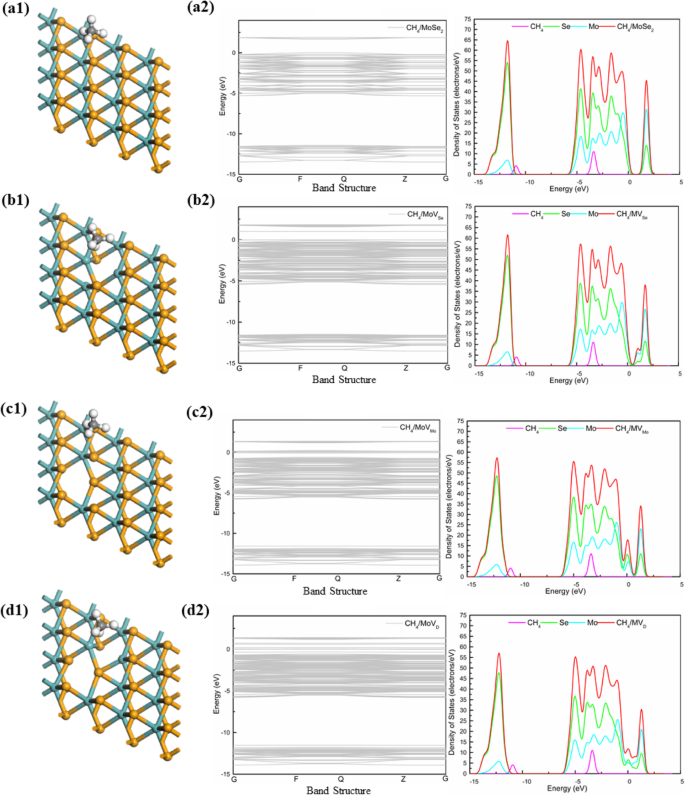
Density of States Analysis
For pristine MoS₂, the band gap is 1.94 eV, decreasing to 0.209 eV for the divacancy. DOS plots show minimal overlap between CH₄ states and the MoS₂ conduction band, confirming physisorption. MoSe₂ displays a smaller band gap (1.68 eV pristine, 0.024 eV divacancy) and similar physisorption behavior. MoTe₂ exhibits the most pronounced changes: the Mo‑vacancy turns the material metallic, and the divacancy retains a small gap (0.316 eV) while showing the largest CH₄ DOS near the Fermi level.
Conclusions
DFT calculations reveal that methane interacts weakly with MoX₂ monolayers, indicative of physisorption. Defect engineering—especially the divacancy in MoTe₂—substantially enhances adsorption energy, charge transfer, and modifies the electronic structure. MV_D(MoTe₂) combines a favorable band gap with strong methane binding, positioning it as a superior candidate for methane sensor development.
Availability of Data and Materials
All data are fully available without restriction.
Nanomaterials
- Nanoparticle-Enhanced Wormlike Micellar System: Design, Rheology, and Mechanistic Insights
- First‑Principles Study of Small‑Molecule Adsorption on Penta‑Graphene for Gas‑Sensing Applications
- First‑Principles Insights into Transition‑Metal Adsorption on Black Phosphorene: Implications for Catalysis and Spintronics
- Gradient Nanomechanical Properties of Fluorosed Enamel: Implications for Restorative Material Selection
- Hexagonal Boron Arsenide as a Highly Sensitive SO₂ Gas Sensor: A First‑Principles Study
- Penta‑Graphene: A Next‑Generation NOx Gas Sensor with Superior Sensitivity
- Rh-Doped MoTe₂ Monolayer: A Powerful DFT-Validated Sensor and Scavenger for SF₆ Decomposition Products
- Theoretical Insights into SF6 Decomposition Product Adsorption on ZnO-Modified C3N Nanosheets
- Al- and P-Doped WS₂ Sensors: Enhanced Detection of NO, NO₂, and SO₂ via First-Principles Analysis
- Tuning Magnetic Properties of Janus WSSe Monolayers via Transition Metal Adsorption